

Short versus Long Reads
Here we look at the effect of read length on performance. Notable changes are:
- Longer reads are faster to align than 36bp reads from the Illumina GA
- The performance improvement is even higher if we consider base pairs aligned per second rather than reads per second.
- Longer single end reads results in an increase in the number of reads aligned to unique locations.
Test Conditions
10,000 Paired end reads generated by ”maq simulate” with a mutation rate of 0.01 (1 mutation per 100 bases which is higher than typical SNP rates) from human chr X and with simulated qualities generated from actual 72 base pair reads. Reads of 36, 48, 60 & 72bp were generated and tested. For single end tests the two paired end files were combined to generate 20,000 single end reads. Reads were aligned against the full human genome at default thresholds.
Default thresholds depend on the size of the genome being searched and the information content of the read. Low quality bases decrease the information content and the default threshold. Typical values for single end reads in this test were:
| Read Length | Low Threshold | Typical Threshold | High Threshold |
|
36
|
80
|
100
|
110
|
|
48
|
150
|
165
|
175
|
|
60
|
190
|
230
|
250
|
|
72
|
210
|
250
|
250
|
Given that a mismatch at base with a Phred quality score of 30 will score 30 we could, on 72bp reads, have potential to align with up to 8 mismatches, or we could have 12 mismatches at bases with Phred quality of 20.
Typical values for paired end reads were:
| Read Length | Low Threshold | Typical Threshold | High Threshold |
|
36
|
260
|
270
|
280
|
|
48
|
330
|
390
|
410
|
|
60
|
380
|
500
|
500
|
|
72
|
450
|
500
|
500
|
In paired end mode the maximum score for each side of a pair is limited to 254 which is more than 8bp mismatch or an insert delete of up to 14bp at default gap penalties. For paired end reads a threshold of 260 could result in an alignment with one end having an alignment score of 10 and the other end a score of 250 as the threshold applies to the whole alignment rather than being split 50-50 between ends.
Single End Reads

Chart 1. 20,000 single end reads from ChrX aligned against full human genome. Increased read length increases the number of uniquely aligned reads and also decreases processing time when compared to 36bp reads. Alignment rate improved from 8.5Kbp/sec for 36bp reads to 32Kbp/sec for 60 & 72 bp reads.
Sensitivity & Specificity
As this test used simulated reads we ran an analysis of true positive and false positive alignments. For this we used the Maq program from Heng Li at the Sanger Institute.
Alignments were first converted into Maq map format using novo2maq and then maq simustat was used to report counts of false positive and total alignments.
We also aligned the reads using Maq to provide a basic comparison.
Read coverage was insufficient to do analysis of SNP or Indel calls.
 |
 |
||
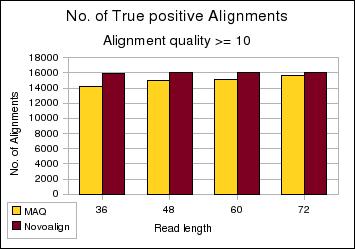
| Chart 2. Shows the number of true positive alignments with an alignment quality score greater than 10 at various read lengths for both Novoalign and Maq. | Chart3. Shows the number of false positive alignments. | ||
Discussion
As read length increases we should expect increased specificity in alignments leading to a drop in false positives and an increase in true positives. However we see a drop in true positives with 72bp reads. This was attributed to reads where the additional length was comprised of low quality bases pushing the alignment score above the Novoalign limit of 254.
Paired End Reads

Chart 4. 10,000 paired end reads from ChrX aligned against full human genome. Increased read length decreases processing time when compared to 36bp reads. Yield of uniquely aligned pairs increases only marginally. Alignment rate improved from 6.5Kbp/sec for 36bp reads to 32Kbp/sec for 60 & 72 bp reads.
Sensitivity & Specificity
Maq simustat was also used to evaluate sensitivity of the paired end alignments.
 |
 |
||
| Chart 5. Shows the number of true positive alignments with an alignment quality score greater than 10 at various read lengths for both Novoalign and Maq. | Chart 6. Shows the number of false positive alignments. | ||
Read Quality

The Human genome index was created with a k-mer size of 14 and a step length of 3.
