

Bisulphite Treated Data with Quality Problem in Read1
Data
45bp paired end reads from lane 3 of Human bisulphite treated DNA. For the following calibration tests we used the first 250,000 reads from the lane.
Early on in the alignment of this data it was noticed that the first read of each pair had a quality problem with overcalls of T starting from base 10 in the reads. The second read of each pair appeared to be OK. The low read quality resulted in two obvious effects: the first was a very low rate of good pair alignments; and the second was very slow run times with the lane taking 48 hrs to prcess on a 16-core server.
Novoalign
9th Jun 2008 – Updated for v2.04.03
Novoalign Command:
- novoalign -h120 -t120 -c12 -u6 -d ..k18s2.human.biseq. -f ..first 250K reads from lane.. -k
|
Uncalibrated
|
Calibrated
|
||
# Paired Reads: 250000 # Pairs Aligned: 123109 # Aligned: 316564 # Unique Alignment: 275463 # Gapped Alignment: 1684 # Elapsed Time: 8782,985s |
# Paired Reads: 250000 # Pairs Aligned: 168968 # Aligned: 373155 # Unique Alignment: 327172 # Gapped Alignment: 1629 # Elapsed Time: 2712.802s |
||
Calibration improved yield of aligned pairs by 37%, increased unique alignments by 19% and run time was cut by 69%.
Alignment Scores
The effects can also be seen in histograms of the number of alignments as a function of the alignment score:
| Uncalibrated | Calibrated | ||
 |
 |
||
The number of alignments to Read 1 of the pairs has increased and the mean alignment score has reduced from 52 to 38. The alignment score reduced because the quality of the bases was reduced and hence there was a lesser penalty for the mismatches. The number of alignments went up because more alignments could be found within the score threshold of 120.
Quality Profile
The quality profiles for the two reads are shown below. Called quality is calculated from the expected number of mismatches given the base qualities over the total calls. Actual base quality is the total number of mismatches over the total calls with both scaled using formula -10log(Perr).

Read 1 quality drops dramatically after base 12 and read 1 actual quality is lower than called quality after base 14. By contrast, actual base qualities on read 2 are higher than the called quality after base 14.
Q40 Calls
The following charts look at Q 40 base calls and the calibrated alignment penalties across the length of the reads. Note that this is bisulphite treated DNA and that it is being aligned against insilico bisulphite treated genomes. As a result there are few T>C mismatches on Read 1 and few G>A mismatches on read2.
|
Base Called
|
Read 1
|
Read 2
|
|
|
A
|
 |
 |
|
|
C
|
 |
 |
|
|
G
|
 |
 |
|
|
T
|
 |
 |
|
The quality of base calls for G & T , as reflected by the calibrated alignment penalty, drops progressively across the read length of Read 1. The second reads of each pair show more consistent read quality.
Quality along the Reads
|
Base
|
Call
|
Read 1
|
Read 2
|
|
0
|
A
|
 |
 |
|
0
|
C
|
 |
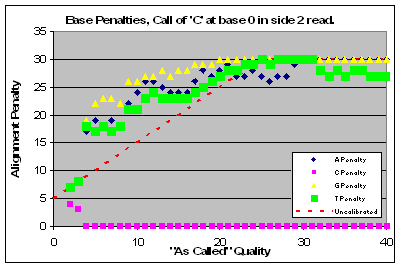 |
|
0
|
G
|
 |
 |
|
0
|
T
|
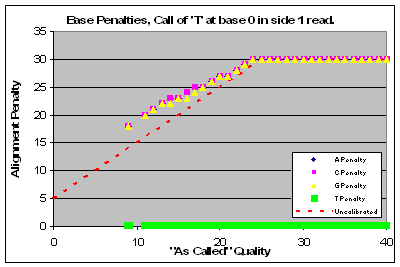 |
 |
|
20
|
A
|
 |
 |
|
20
|
C
|
 |
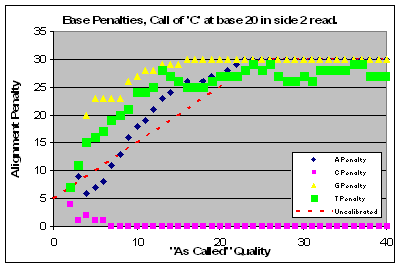 |
|
20
|
G
|
 |
 |
|
20
|
T
|
 |
 |
|
40
|
A
|
 |
 |
|
40
|
C
|
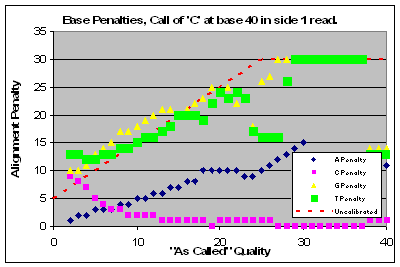 |
 |
|
40
|
G
|
 |
 |
|
40
|
T
|
 |
 |
Read 1 of this lane had a major problem with qualities that affected base calls of C & T and to a lesser extent G’s. Base calls of A’s were of higher quality than as called.
On Read 2 actual qualities were generally higher than the called qualities.
